#特殊的一顿饭##2023国际罕见病日#
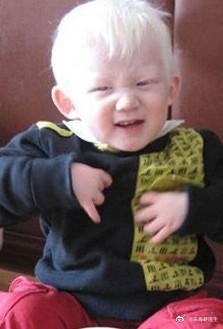
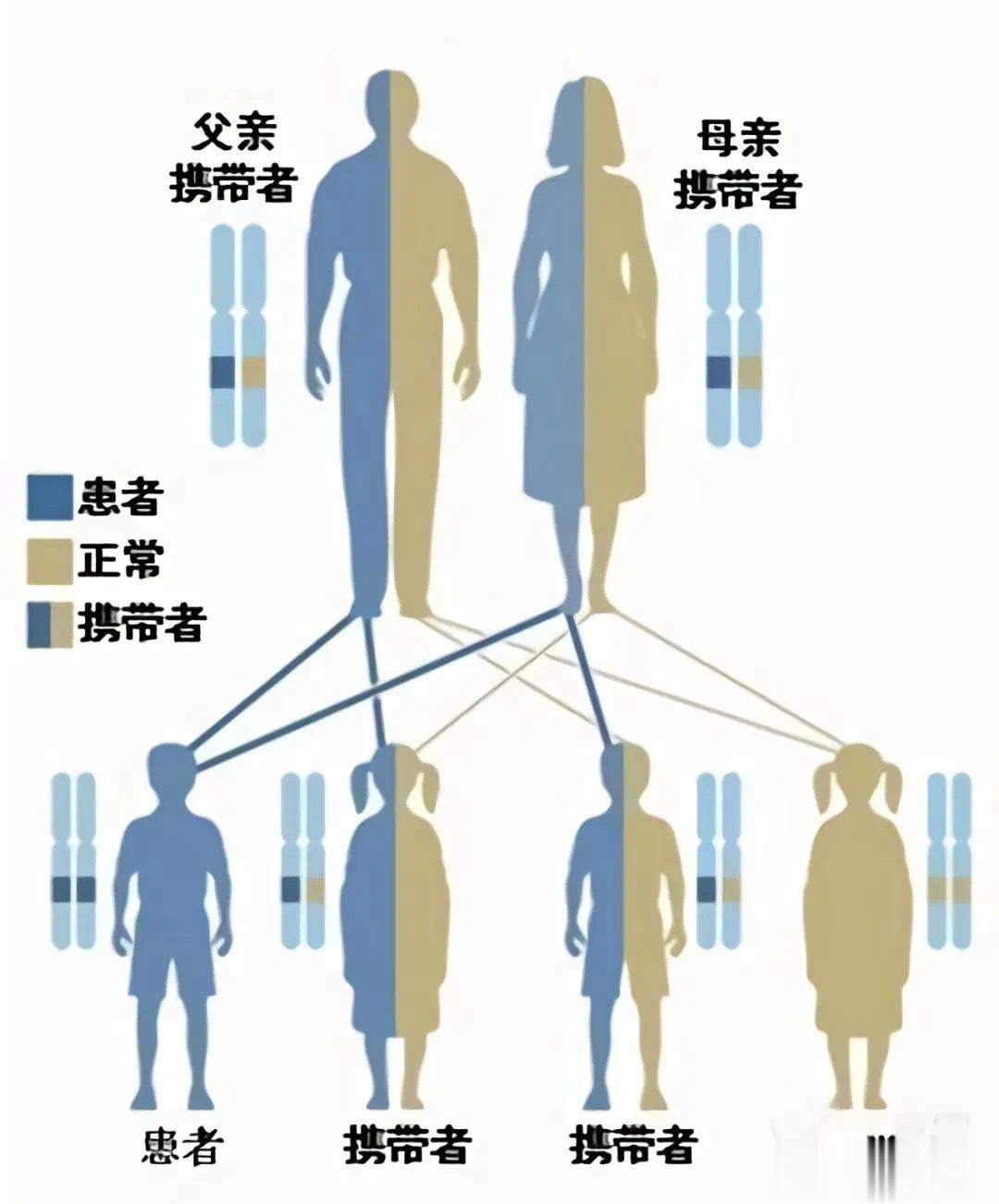
【苯丙酮尿症患者为什么不能吃常规的饮食?】苯丙酮尿症(PKU)是一种遗传代谢性疾病,罕见病之一,发病率在万分之一左右。全国约有10万人患有此病。它是一种体内芳香族氨基酸(苯丙氨酸)代谢途径中酶的缺陷引起的疾病。如果不进行早期干预和治疗,会导致不可逆的智力障碍及其他临床症状。头发黄、皮肤白、“鼠尿味”这是PKU婴儿期的典型外部特征。#儿童健康守护者计划#
笨丙氨酸是人体的必需氨基酸之一。蛋白质的主要成分就是氨基酸, 所以需要控制蛋白质饮食的摄入。该类患儿一辈子都要吃一种特殊的食谱,否则将面临智力损伤、痴呆甚至死亡的风险。由于不能吃常规的高蛋白食物,因此也被人称之为“不食人间烟火的人”。
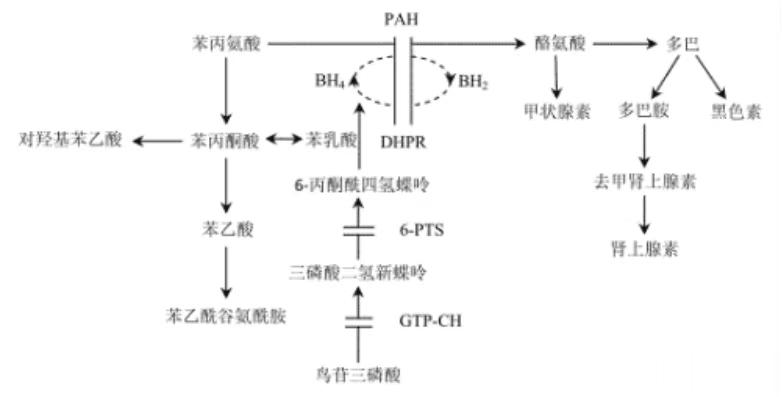
患儿主要是缺乏苯丙氨酸羟化酶,不能将苯丙氨酸转化为酪氨酸。因此,苯丙氨酸在血、脑脊液、各种组织和尿液中的浓度增高,同时产生大量苯丙酮酸、苯乙酸、苯乳酸和对羟基苯乙酸等旁路代谢产物自尿中排出,高浓度的苯丙氨酸及其旁路代谢产物可导致脑细胞损伤。此外,因酪氨酸来源减少,致使甲状腺素、肾上腺素和黑色素等合成不足。
苯丙氨酸浓度升高会抑制大分子中性氨基酸(large neutral amino acid, LNAA)向脑内转运。LNAA减少被认为可抑制蛋白质和神经递质的合成,从而导致多巴胺和5-羟色胺浓度不足。
还有少部分患儿是由于四氢生物蝶呤(BH4)缺乏。BH4是苯丙氨酸、酪氨酸和色氨酸等芳香氨基酸在羟化过程中所必需的共同的辅酶,BH4缺乏时不仅苯丙氨酸不能氧化成酪氨酸,而且造成多巴胺、5-羟色胺等重要神经递质的合成受阻,加重了神经系统的功能损害。这种类型的PKU的临床症状更严重,治疗更难。
苯丙酮尿症在新生儿期并无症状,但是可以通过新生儿足底采血筛查发现。如果新生儿筛查未能检出PKU,则PKU的发病将会较为隐匿,直到婴儿早期才引起症状。
在未经治疗的患者中,PKU的特点为不可逆性智力障碍、癫痫发作、行为异常、小头畸形以及皮肤的异常。宝宝刚出生的时候,肤色正常,生后数月,由于黑色素合成不足,头发、皮肤和虹膜色素变浅,皮肤湿疹多见。其他的表现包括步态、坐姿和站姿异常。由于苯乙酸浓度增加,宝宝的身体和尿液可能有“鼠”臭味。
若不采取膳食限制,随着膳食中的苯丙氨酸暴露不断增加,儿童早期髓鞘形成过程中的认知损害会加重。
BH4缺乏的患儿由于神经递质(多巴胺、肾上腺素、去甲肾上腺素和5-羟色胺)的产生减少,通常会在婴儿期出现你写的皮肤症状和/或进行性神经功能恶化。未治疗的患者常在1岁前死亡。
膳食限制是PKU治疗的主要措施
PKU的主要治疗方法仍是饮食限制苯丙氨酸。膳食治疗需食用医疗食品,包括不含苯丙氨酸的蛋白替代物,其可满足约75%的蛋白质需求(苯丙氨酸除外)。
医疗食品中的蛋白质来源可能为糖巨肽(GMP)。GMP是干酪乳清中的一种天然蛋白质,含有少量的苯丙氨酸并补充了若干大分子中性氨基酸(LNAA)。多项研究已证实,PKU患者采用GMP饮食治疗具有有效性和适口性。
在经验丰富的代谢营养师监督指导下,PKU婴儿可以实行一部分母乳喂养,并搭配不含苯丙氨酸的配方奶粉喂养。母乳喂养的占比通常限制在25%左右,具体取决于疾病的严重程度。经母乳摄取苯丙氨酸时,必须考虑其每日允许摄入量。
蛋白替代品的适口性差可对饮食依从性产生不利影响,尤其是对于年龄较大的儿童。患者所需的苯丙氨酸量通过少量天然蛋白来提供。极少数患者可能需要补充酪氨酸。
对于存在PKU且血苯丙氨酸浓度>6mg/dl (360μmol/L)的婴儿,应尽早开始治疗,通常在出生后1周内开始。
低苯丙氨酸饮食原则是使苯丙氨酸的摄入量既能保证生长和代谢的最低需要,又要避免血中含量过高。
婴儿主要给予低苯丙氨酸奶粉,待血苯丙氨酸浓度降至理想范围时可逐渐添加天然食品。因母乳中苯丙氨酸含量仅为牛奶的1/3,故为首选。
幼儿以淀粉类、蔬菜、水果等低蛋白饮食为主。低苯丙氨酸饮食治疗至少应持续到青春期,但终身治疗对患者更为有益。