
成果简介
人类的生命依赖于过氧化氢(H2O2),这是一种至关重要的化学物质。H2O2可以在原子分散的催化剂上通过双电子氧还原反应(2e−ORR)产生,这使其成为传统蒽醌工艺的有效替代方案,而活性位点的轴向配位调节是提高2e−ORR选择性的一种有效手段。
在此,福州大学丁开宁、不来梅大学郭翔宇等人设计了20种单原子位点,其具有通过轴向配位R(R=Cl,Br)功能化的M–N4–C结构。计算方法基于DMol3模块,作者采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)交换关联泛函来进行密度泛函理论计算,并采用DFT+D2的经验校正来处理范德华(vdW)作用。
作者使用密度泛函半核赝势(DSPP)来研究过渡金属的核心电子,并使用双数值加极化(DNP)基组来描述价电子。在整个计算中,作者将能量、力和位移的收敛标准分别设定为1×10−5 Ha、0.002 Ha/Å和0.005Å,并且自洽场(SCF)计算的迭代标准设置为10−6 Hartree。作者使用类导体屏蔽模型(COSMO)来对实际水溶液进行模拟,其中水的介电常数为78.54。作者分别使用5×5×1和7×7×1的K点网格进行几何优化和电子性质计算,并使用Hirshfeld方法来计算电荷转移。
结果与讨论
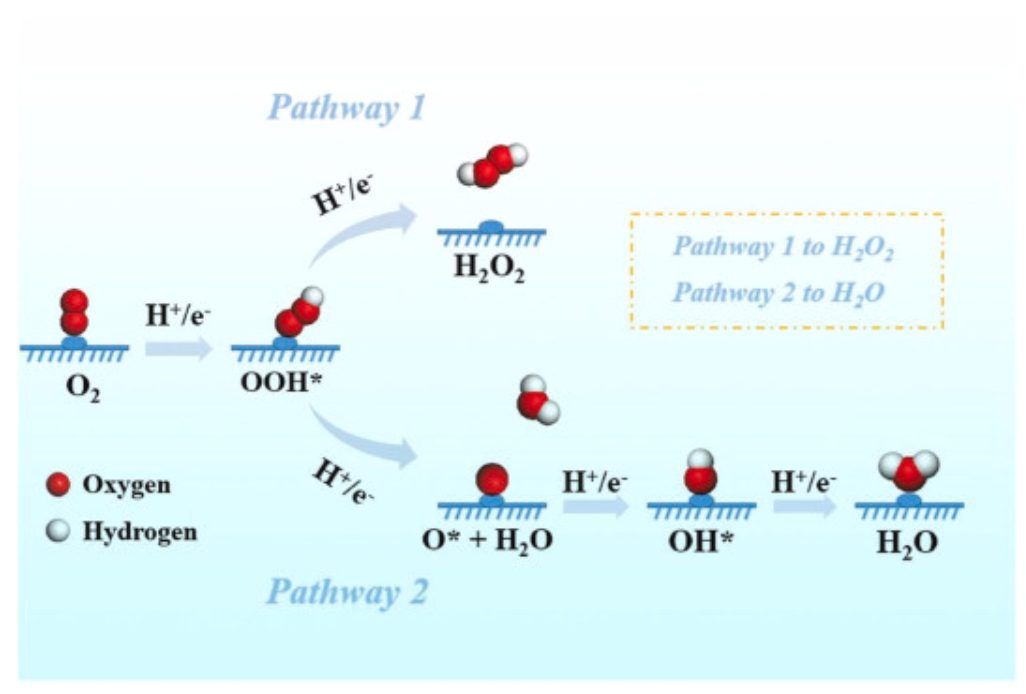
图1 ORR途径
如图1所示,ORR涉及两个竞争过程,分别通过2e−途径将O2还原为H2O2,以及通过4e−途径将O2还原为H2O。在热力学上,该反应倾向于产生H2O,并且它是通过2e−途径合成H2O2的巨大障碍。为了产生H2O2,需要OOH*到H2O2的能垒要低于OOH*氢化为O*的能垒,这意味着中间体O*的吉布斯自由能变化值(ΔGO*)必须大于3.52eV,因此ΔGO*>3.52eV成为判断2e−ORR催化剂的基础。
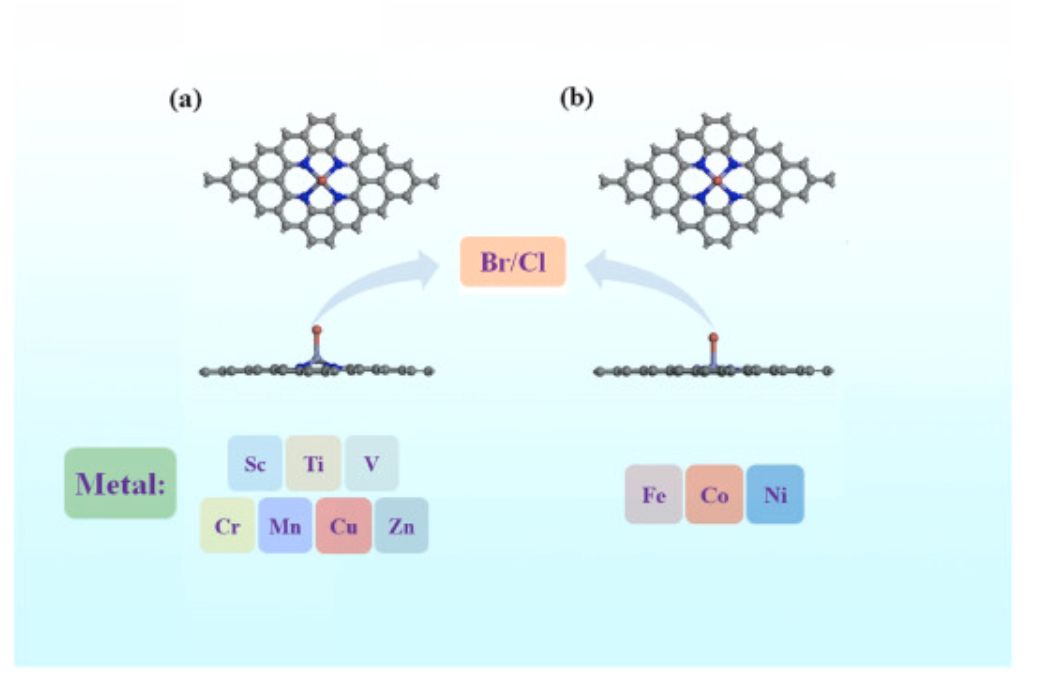
图2 催化剂模型结构
如图2所示,受不同金属原子尺寸和轴向配体的影响,包括Sc、Ti、V、Cr、Mn、Cu和Zn在内的金属原子在掺杂后倾向于从石墨烯表面突出,而Fe、Co和Ni可以很好地嵌入石墨烯晶格中,呈现出准平面的M–N4–C构型。
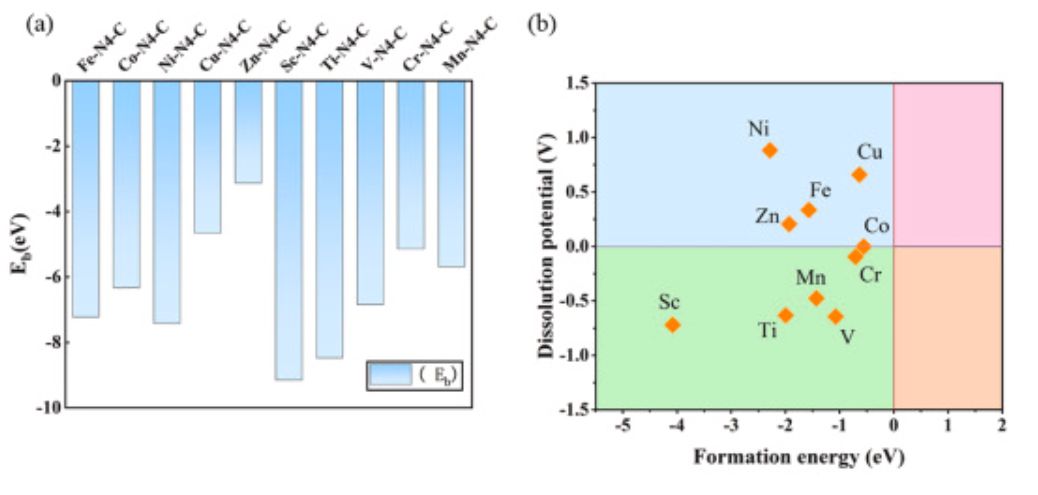
图3 结合能、形成能与溶解势
如图3a所示,负的结合能表明金属和基质之间可以良好结合,表明这些结构具有较好的稳定性。如图3b所示,基于形成能和溶解势,作者将这些模型分为四种类型,分别对应于图中的蓝色、粉红色、绿色和橙色区域。位于蓝色区域的M–N4–C(M=Zn、Fe、Cu和Ni)不仅具有良好的热力学稳定性,而且溶解势大于零,这表明这些材料可以有效地抑制金属原子溶解,并具有优异的电化学稳定性。因此,作者选择了这四种SAC来构建Cl和Br的轴向配位,从而产生了八种结构。

图4 AIMD模拟
如图4所示,在AIMD中,Zn–N4–Br的几何构型得到了有效维持,并且在500K下模拟10ps时,温度和能量的振动范围都比较窄,这表明Zn–N4-Br催化剂在反应中具有较高的热力学稳定性。此外,作者在声子色散谱中没有观察到明显的虚频,这表明它具有优异的动力学稳定性。

图5 Zn、Fe、Cu和Ni基催化剂的ΔG(O*)值
在所考虑的12种SAC中,Fe–N4–Cl、Fe–N4-Br、Zn–N4-Cl、Ni–N4-Cl和其他三种没有轴向原子配位的Cu、Zn和Ni基催化剂的ΔG(O*)值<3.52eV,表明它们对H2O2合成的选择性较差。而剩下的四种SAC满足选择性标准(ΔG(O*)>3.52eV),其中包括Cu–N4–Cl、Ni–N4-Br、Cu–N4-Br和Zn–N4—Br。此外,在没有轴向配位的情况下,Ni–N4–C和Cu–N4-C倾向于产生H2O而不是H2O2,这表明金属原子的配位环境在SAC的催化选择性中起着重要作用。

图6 势能面
如图6所示,在U=0.0 V时,ORR的所有基元步骤都是放热的,这意味着反应很容易发生。在反应的基元步骤中,电势决定步骤(PDS)是具有最高ΔG值和最低阶跃下降的步骤,而理论极限电势的公式为UL=ΔGPDS/e可,所以Cu–N4–Br、Cu–N4-Cl、Zn–N4-Br和Ni–N4-Br的理论极限电势分别为0.55 V、0.63 V、0.65 V和0.27 V。
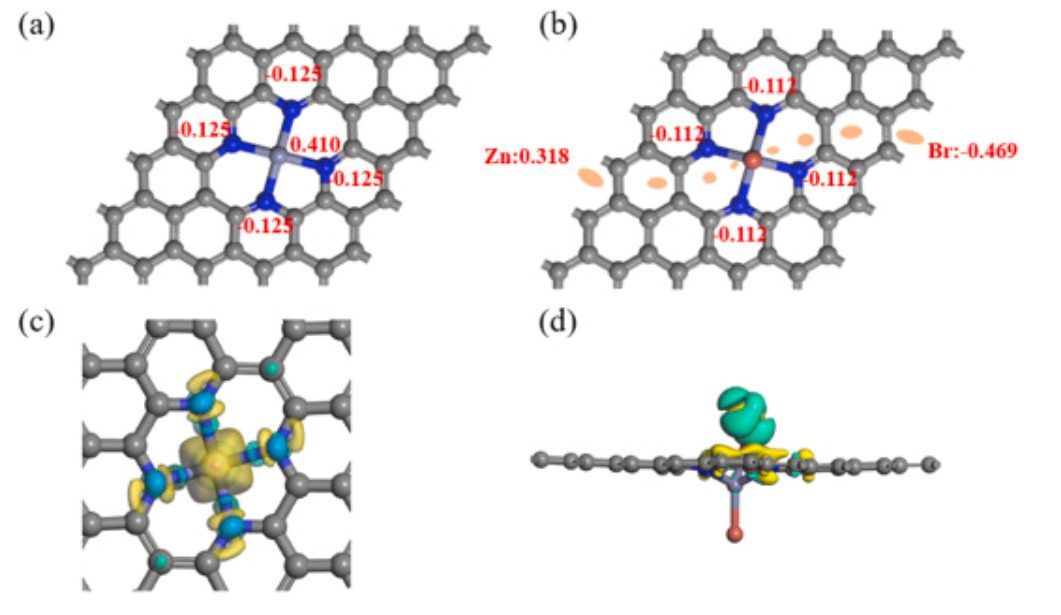
图7 电荷布居和差分电荷密度
如图7所示,当轴向配体Br原子引入Zn–N4–C的垂直方向时,金属Zn上的Hirschfeld电荷从0.410下降到0.318(图6a-b),表明Zn原子失去了更多的电子。此外,当OOH*物种吸附在Zn–N4–Br/石墨烯上时,差分电荷密度图(图7d)表明Zn原子和OOH*之间有相当大的电荷转移(从Zn原子到OOH*的电子转移)。因此,O–O键被Zn单原子成功活化,这使得OOH*更容易通过氢化产生H2O2。

图8 PDOS
如图8所示,当O2以自旋三重态吸附在Zn–N4–Br催化剂上时,O2的π*和π轨道与Zn的d轨道重合。金属Zn原子的空d轨道接受来自O2成键轨道的电子,同时,当自旋向下的π*轨道峰值从费米能级以上降低时,被占据的Zn d轨道将电子回馈到O2 2π反键轨道。当O2分子被氢化为OOH*时,OOH*中的O–O键被适度活化,因为Zn的3d轨道与O2的2p轨道重叠,这意味着Zn–N4–Br上的ORR倾向于遵循2e−路径。
结论与展望
作者通过密度泛函理论(DFT)计算发现,与原始的M–N4–C结构不同,Cl–Cu–N4和Br–Zn–N4具有优异的2e−ORR催化效率,过电位分别为0.07和0.05V。此外,中间体O*的吉布斯自由能超过3.52eV,表明竞争性4e−ORR反应被显著抑制。电子分析表明,Zn–N4–Br中的轴向Br可以优化Zn中心的3d轨道,增强O2在Zn位点的吸附和活化,从而降低ORR势垒,加速ORR动力学。
文献信息
Qianqian Liu et.al Axial halogen coordinated metal-nitrogen-carbon moiety enables efficient electrochemical oxygen reduction to hydrogen peroxide International Journal of Hydrogen Energy 2023
