
英文原题:Machine-Learned Electronically Excited States with the MolOrbImage Generated from the Molecular Ground State

通讯作者:任咏华,香港大学
作者:陈自勇,任咏华
研究背景
与激发态性质相关的判据对于发光材料的筛选与设计有指导意义。例如,高效率的热活化延迟荧光发光分子往往具有较小的单–三重态分裂能(ΔES-T)。虽然密度泛函理论已经广泛应用于激发态计算,其成本还是远大于对分子基态的计算,对三重态激发能也有较大的预测误差。而要包含激发态的电子相关效应,则需要进一步增加成本。
因此,使用机器学习考虑电子相关效应,对激发态性质进行高精度地预测,成为了另一个实用的方案。相比对分子基态性质的大量研究,机器学习在激发态方面的应用还相当有限。将基态计算得到的分子轨道,与不同的量子算符结合,可以生成各式的电子积分矩阵。有趣的是,矩阵本身可以视作一个二维的分子轨道图像(MolOrbImage)。受到卷积神经网络(CNN)在计算机视觉领域巨大成功的启发,我们相信将CNN与MolOrbImage结合对于激发态性质的预测也会达到理想的准确度。
快讯亮点
以电子相关方法[ADC(2)]为基准,通过构建以卷积层为主导的神经网络,本项工作对分子激发态性质进行了准确预测,包括:
(1)对第一单重态(S₁)和三重态(T₁)能量,平均绝对误差优于文献报道;
(2)单重态跃迁振子强度得到了定性预测;
(3)对于不在训练集范围内的分子,模型展现了一定的可迁移性。
内容介绍
本文选取了40000个QM9分子(最多包含9个重原子,包括碳,氮,氧和氟),在ADC(2)/cc-pVTZ水平下计算了激发能和振子强度,以用作训练和测试集。结果表明,对S₁(图1a)和T₁(图1b)激发态能量预测的误差呈现正态分布,且平均绝对误差也随着训练集中分子数目(Ntrain)的增加而稳步下降。整体结果优于先前的文献报道。对于比较困难的振子强度,预测的准确性也随着Ntrain的增大而增加,fS1在Ntrain=32000时相关系数r达到了0.86。虽然我们的模型不会直接学习激发态间的分裂能,这些分裂能可以间接地通过绝对能量相减得出。本文展示了三个对分子设计具备指导意义的能差(图1d-f),其中的ΔET1-T2和本课题组最近提出的热刺激延迟磷光(TSDP)紧密相关。对于三个能差,预测结果和参考值间均显示了非常强的相关性(r>0.9)。
一个有实际应用价值的机器学习模型也需要能够对不在训练集中的分子做出合理预测。本文通过外推集分子(图2a)展示了模型优异的迁移性。对内插分子(图2b,2d),特别是ET1,模型预测结果明显的优于基于密度泛函理论的计算值。到了外推集,模型的预测平均绝对误差只是稍微增加,而且相关系数也大于0.95(图2c,2e)。我们相信,基于分子轨道的MolOrbImage,可以获取外推分子的重要特征,从而使模型具有强大的迁移性。
综上所诉,本文基于卷积神经网络和分子轨道图像,在基态计算的成本上可以得到含有电子相关效应的激发态性质。该工作不仅是对机器学习激发态研究的扩充,也可以为发光功能材料的筛选和设计提供理论指导。
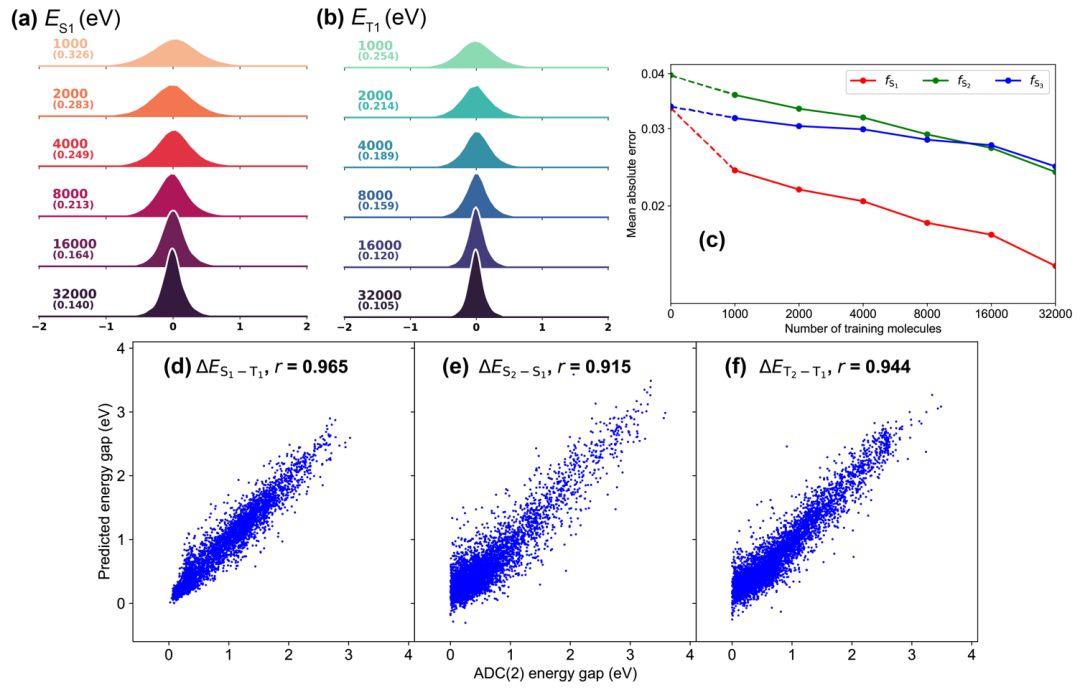
图 1. 模型的预测结果:第一(a)单重态和(b)三重态激发能的误差分布随训练集体量的变化,平均绝对误差在括号中给出;(c) 单重态振子强度的学习曲线;(d) ΔES1-T1,(e) ΔES2-S1 和(f) ΔET2-T1的预测值与参考值,r为相关系数。

图 2. 模型的迁移能力:(a)我们选取了测试集中的前100个分子作为內插集,并通过对内插分子的改造得到了外推集。在外推集中,分子可以含有第三周期元素,或者重原子数大于9。(b,d) 内插集和(c,e)外推集的机器学习结果和基于B3LYP的计算结果也进行了对比。
J. Phys. Chem. Lett.2023, 14, 7, 1955–1961
Publication Date: February 14, 2023
