自动注释与上面的方法不同,它们基于不同的原理,有时需要预定义的标记集,有时在预先存在的完整 scRNA-seq 数据集上进行训练。生成的结果可能具有不同的质量。因此,将这些方法视为注释过程的起点而不是终点非常重要。
自动生成的注释的质量可能会有所不同。更具体地说,注释的质量取决于:
●选择的分类器类型:以前的基准研究表明,不同类型的分类器通常表现相当,基于神经网络的方法通常不会优于通用模型,例如支持向量机或线性回归模型。
●训练分类器所依据的数据的质量。如果训练数据没有得到很好的注释或以低分辨率进行注释,则分类器将执行相同的操作。同样,如果训练数据和/或其注释有噪声,则分类器的性能可能不佳。
您自己的数据与训练分类器所依据的数据的相似性。例如,如果分类器是在 drop-seq 单细胞数据集上训练的,您的数据是 10X 单细胞核而不是单细胞 drop-seq,这可能会降低注释的质量。与在单个数据集上训练的分类器相比,在跨数据集图集(包括各种数据集)上训练的分类器可能会提供更强大、质量更好的注释。一个例子是在人类肺细胞图谱上训练的 CellTypist分类器,其中包括 14 个不同的肺数据集。与在单个肺数据集上训练的模型相比,该模型在新的肺数据上的性能可能更好。
使用预先训练的分类器来注释数据有几个重要优势。
●首先,这是一种快速简便的数据注释方法。注释不需要下载或预处理训练数据,有时只涉及将数据上传到在线网页。
●其次,这些方法不像手动注释那样依赖于将数据分区到集群中。
●第三,预先训练的分类器使您能够直接利用以前研究的知识和信息,例如高质量的注释。
●最后,使用此类分类器可以帮助协调整个字段的单元格类型定义,从而为在字段范围内就这些定义达成共识扫清道路。
类型
基于标记基因的分类器
一类自动细胞类型注释方法依赖于一组预定义的标记基因。根据这些标记基因的表达水平将细胞分为细胞类型。此类方法的示例是Garnett和CellAssign。这些模型所基于的标记基因集越健壮和可推广,模型的性能就越好。但是,与其他模型一样,它们可能会受到模型训练所依据的数据与需要标记的数据之间与批次效应相关的差异的影响。与基于较大基因集的模型相比,这些方法的优势之一是它们更加透明:我们知道根据哪些基因进行分类。
基于更广泛的基因集的分类器
到目前为止讨论的方法只使用数据中检测到的基因的一小部分:通常每种细胞类型仅使用一组 1 到 ~10 个标记基因。另一种方法是使用分类器,该分类器将更大的基因集(数千个或更多)作为输入,从而更多地利用 scRNA-seq 数据的广度。此类分类器在先前注释的数据集或图集上进行训练。这些例子是CellTypist和Clustifyr。
CellTypist代码展示
这里我们需要原始数据并进行对数平移变换的标准化:
adata_celltypist = adata.copy() # make a copy of our adataadata_celltypist.X = adata.layers["counts"] # set adata.X to raw countssc.pp.normalize_per_cell( adata_celltypist, counts_per_cell_after=10**4) # normalize to 10,000 counts per cellsc.pp.log1p(adata_celltypist) # log-transform# make .X dense instead of sparse, for compatibility with celltypist:adata_celltypist.X = adata_celltypist.X.toarray()
下载注释集:
models.download_models( force_update=True, model=["Immune_All_Low.pkl", "Immune_All_High.pkl"])
让我们尝试Immune_All_Low 和 Immune_All_High 模型(它们注释免疫细胞类型更精细的注释级别(低)和更粗糙的注释级别(高)):
model_low = models.Model.load(model="Immune_All_Low.pkl")model_high = models.Model.load(model="Immune_All_High.pkl")

两种模型
当然我们也可以直接查看:
model_high.cell_typesmodel_low.cell_types
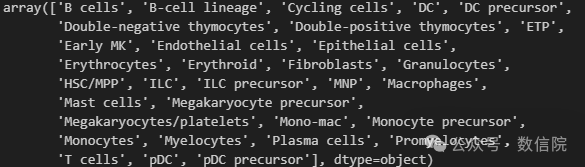
low cell
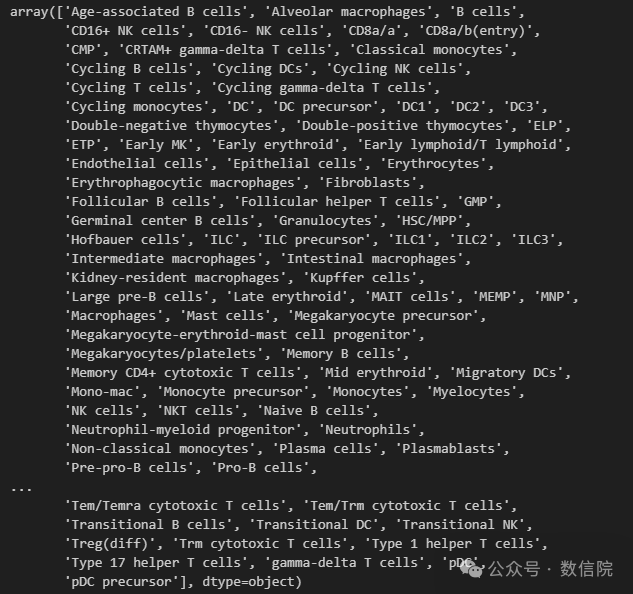
high group
我们粗略的运行一下:
predictions_high = celltypist.annotate( adata_celltypist, model=model_high, majority_voting=True)
计算过程图
我们赋值给一个新的对象:
predictions_high_adata = predictions_high.to_adata()
并添加到原始对象中:
这样,adata 对象中将会新增两个列,分别表示每个细胞的预测标签和对应的置信度分数。这些列可以用于后续的分析、筛选或可视化。
adata.obs["celltypist_cell_label_coarse"] = predictions_high_adata.obs.loc[ adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_coarse"] = predictions_high_adata.obs.loc[ adata.obs.index, "conf_score"]
接下来我们换另一个集:
predictions_low = celltypist.annotate( adata_celltypist, model=model_low, majority_voting=True)
赋值给新对象:
predictions_low_adata = predictions_low.to_adata()
添加到adata中:
adata.obs["celltypist_cell_label_fine"] = predictions_low_adata.obs.loc[ adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_fine"] = predictions_low_adata.obs.loc[ adata.obs.index, "conf_score"]
然后我们画图:
sc.pl.umap( adata, color=["celltypist_cell_label_coarse", "celltypist_conf_score_coarse"], frameon=False, sort_order=False, wspace=1,)


结果图
可以看到,结果很差。
原因是什么呢?
原因是数据集是有关肾脏的整体转录数据,而自动注释的包仅仅针对于免疫细胞,因此在用于肾脏整体单细胞转录组的时候,注释结果便会不准确。相反, CellTypist更多针对于免疫细胞,在用于骨髓等相关数据的时候可能会有更好的结果。因此我们更换一个数据重新进行分析。
读入数据:
adata = sc.read( filename="s4d8_clustered.h5ad", backup_url="https://figshare.com/ndownloader/files/41436666",)
我们重复上述代码:
models.download_models(force_update=True, model=["Immune_All_Low.pkl", "Immune_All_High.pkl"])model_low = models.Model.load(model="Immune_All_Low.pkl")model_high = models.Model.load(model="Immune_All_High.pkl")predictions_high = celltypist.annotate(adata_celltypist, model=model_high, majority_voting=True)predictions_high_adata = predictions_high.to_adata()adata.obs["celltypist_cell_label_coarse"] = predictions_high_adata.obs.loc[adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_coarse"] = predictions_high_adata.obs.loc[adata.obs.index, "conf_score"]predictions_low = celltypist.annotate(adata_celltypist, model=model_low, majority_voting=True)predictions_low_adata = predictions_low.to_adata()adata.obs["celltypist_cell_label_fine"] = predictions_low_adata.obs.loc[adata.obs.index, "majority_voting"]adata.obs["celltypist_conf_score_fine"] = predictions_low_adata.obs.loc[adata.obs.index, "conf_score"]sc.pl.umap(adata,color=["celltypist_cell_label_coarse", "celltypist_conf_score_coarse"],frameon=False,sort_order=False,wspace=1,)

cell_label

conf_score
我们再看这个注释图,就会好很多。
更细致的如下:
sc.pl.umap( adata, color=["celltypist_cell_label_fine", "celltypist_conf_score_fine"], frameon=False, sort_order=False, wspace=1,)
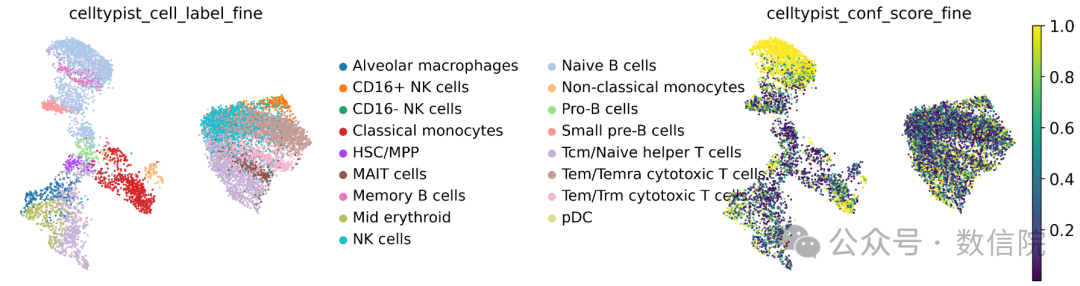
标签和打分图
查看观察到的单元格类型相似性是否符合我们的期望:
sc.pl.dendrogram(adata, groupby="celltypist_cell_label_fine")

图例
该树状图部分反映了对细胞类型关系的先验知识(例如 B 细胞主要聚集在一起),但我们也观察到一些意想不到的模式:Tcm/Naive 辅助性 T 细胞与红细胞和巨噬细胞聚集,而不是与其他 T 细胞聚集。这是一个危险信号!可能,Tcm/Naive 辅助性 T 细胞注释是错误的。需要手动注释进行矫正。